 Computational Methods for Paleogenomics and Comparative Genomics
Computational Methods for Paleogenomics and Comparative Genomics


|
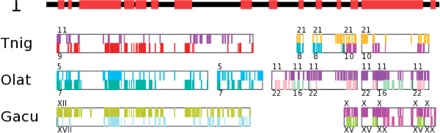
The local approach principle was first developed for the case of
one-to-one orthologous genomic markers (genes, synteny blocks,
...). Its principle, that relies on the combinatorial concept of
Consecutive Ones Property of Binary Matrices, was outlined in
[PLoS
Comput. Biol. 2008] and implemented in the
software ANGES
[Bioinformatics
2012]. Recently, this approach has been extended to handle
duplicated markers
[BMC
Bioinformatics 2012, SFU
PhD thesis, 2015]. Our algorithms and softwares have been applied
to several ancestral genomes, including mosquitoes
[Science
2014], yeasts
[JCB 2010],
plants
[Bioinformatics
2011], amniotes
[Bioinformatics
2011].
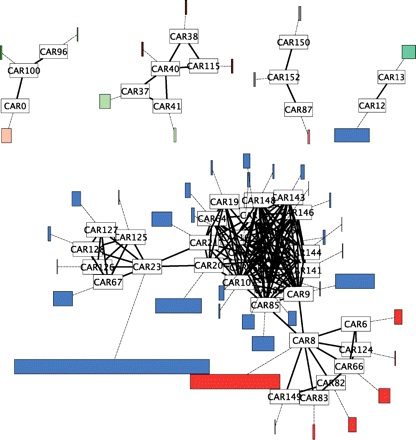
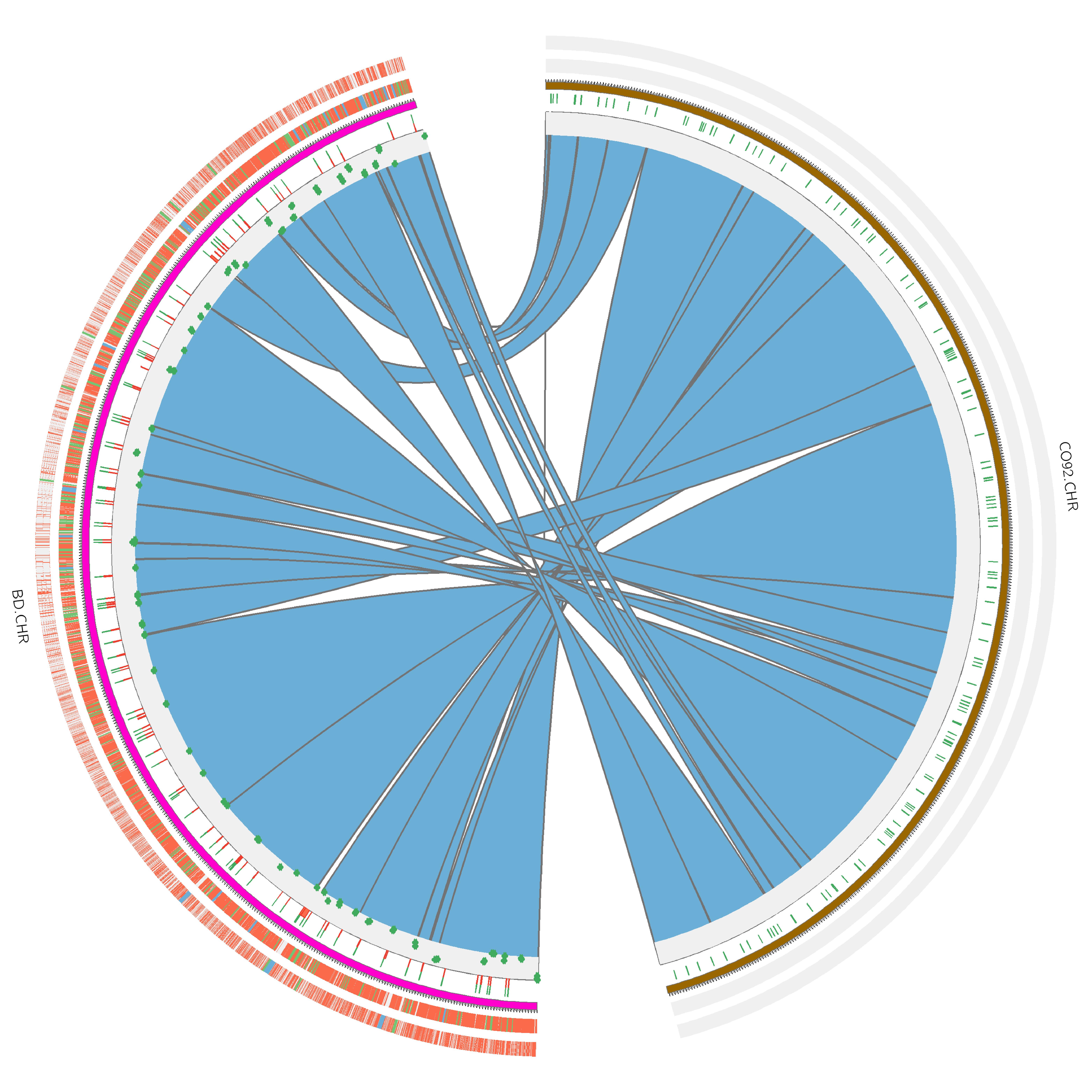
Our work on the small parsimony problem follows from the seminal work of Sankoff and El-Mabrouk that links reconciled gene trees and ancestral genomes (reviewed in [MAGE 2013]). Our main contribution is a probabilistic dynamic programming algorithm for the reconstruction of the evolutionary histories of gene adjacencies, implemented in the software DeClone [biorxiv 2015] that can also apply to scaffold fragmented extant genomes [BMC Genomics 2015]. The most recent direction of research in our lab is the integration of both approaches into a unified global ancestral genome reconstruction pipeline. The recent breakthroughs in ancient DNA (aDNA) sequencing naturally lead us to investigate the problem of applying the principles of the methods we developed to reconstruct ancestral genomes maps to the assembly of aDNA sequencing data. Our first results, on scaffolding the genome of the Black Death agent illustrate the relevance of this approach [Bioinformatics 2013]. Finally, our line of work that aims at using duplicated genes for the reconstruction of ancestral genomes motivated a series of papers on the correction of gene trees [Bioinformatics 2014] and on the reconciliation between gene trees and species trees [TCBB 2012]. |
|
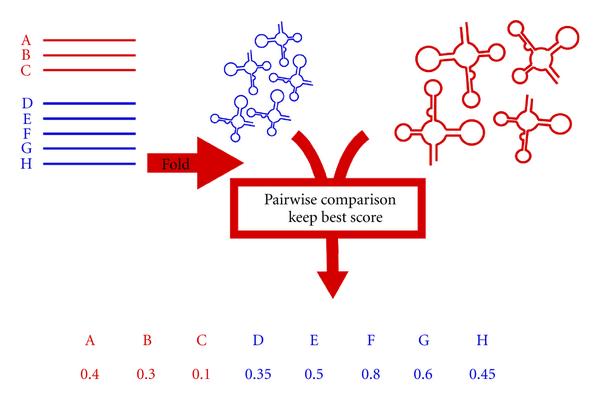
We are mostly interested in developing accurate and efficient
algorithm to compare pairs of RNA secondary structures. Among others, we have
been involved in the development of
the BRASERO benchmark, and in
the design of efficient dynamic programming algorithms for comparing
RNA secondary structures
[TCS 2011].
Our most recent work, in collaboration with Bordeaux University, showed that the chaining approach, widely used in sequence comparison, can be extended very to RNA secondary structures to offer an efficient and accurate method to find structure-sequence homologs in a database of RNA sequences [JDA 2012; JCB 2015]. |